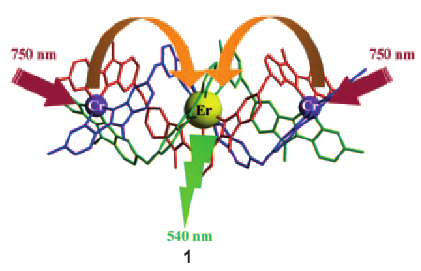-
Optimizing Millisecond Time Scale Near-Infrared Emission in Polynuclear Chrome(III)-Lanthanide(III) Complexes
L. Aboshyan-Sorgho, H. Nozary, A. Aebischer, J.-C.G. Bünzli, , K.R. Kittilstved, A. Hauser, S.V. Eliseeva, S. Petoud and C. Piguet
Journal of the American Chemical Society, 134 (30) (2012), p12675-12684


DOI:10.1021/ja304009b | unige:22645 | Abstract | Article HTML | Article PDF

This work illustrates a simple approach for optimizing long-lived near-infrared lanthanide-centered luminescence using trivalent chromium chromophores as sensitizers. Reactions of the segmental ligand L2 with stoichiometric amounts of M(CF3SO3)2 (M = Cr, Zn) and Ln(CF3SO3)3 (Ln = Nd, Er, Yb) under aerobic conditions quantitatively yield the D3-symmetrical trinuclear [MLnM(L2)3](CF3SO3)n complexes (M = Zn, n = 7; M = Cr, n = 9), in which the central lanthanide activator is sandwiched between the two transition metal cations. Visible or NIR irradiation of the peripheral Cr(III) chromophores in [CrLnCr(L2)3]9+ induces rate-limiting intramolecular intermetallic CrâLn energy transfer processes (Ln = Nd, Er, Yb), which eventually produces lanthanide-centered near-infrared (NIR) or IR emission with apparent lifetimes within the millisecond range. As compared to the parent dinuclear complexes [CrLn(L1)3]6+, the connection of a second strong-field [CrN6] sensitizer in [CrLnCr(L2)3]9+ significantly enhances the emission intensity without perturbing the kinetic regime. This work opens novel exciting photophysical perspectives via the buildup of non-negligible population densities for the long-lived doubly excited state [Cr*LnCr*(L2)3]9+ under reasonable pumping powers.